INTRODUCTION
CRISPR technologies offer unprecedented precision and efficiency in gene editing and therapy. In the context of dominant genetic disorders, it is of paramount importance to target the pathogenic mutation of an allele while preserving the healthy one. Unfortunately, the permissive nature of DNA-gRNA interactions alone in CAS systems is not sufficient to guarantee this requested precision. For this reason, exploiting SNV-derived PAMs (i.e. single nucleotide variants (SNV) leading to the introduction of a Protospacer Adjacent Motif (PAM) at a target genomic site) has emerged as an effective strategy to unlock the full potential of genome editing (György, B. et al., 2019). The AlPaCas (Aligning Patients to Cas) webserver is an automated pipeline for sequence-based identification and structural analysis of SNV-derived PAMs. AlPaCas is a Python-based protocol that, when provided with a gene input (HGNC GeneSymbol, NCBI Gene-ID, VCV-identifiers or raw sequences, can: 1) detect the presence of SNV-derived PAMs; 2) provide a list of available CAS enzymes recognizing the identified PAM(s); 3) propose CAS-engineering mutations to enhance the selectivity of PAM recognition. As output, AlPaCas offers a comprehensive list of SNV-derived PAMs, complete with the original sequences and suitable Cas enzymes for targeted editing. It achieves this by querying an internal SQL database, monthly updated, which minimizes disruptions from each server-side call. The 3D-structures of Cas enzymes, available from PDB or provided by users, are exploited to analyze each residue’s contribution to Cas-PAM interaction. In pursuit of enhancing precise PAM recognition, we apply established protocols on such residues for Evolutionary conservation and ΔΔG computations (Huang, X. et al., 2023; Paiardini, A. et al., 2005) to propose structure-based Cas engineering strategies. In conclusion, the AlPaCas webserver, with its automated pipeline and comprehensive analysis, stands at the forefront of advancing CRISPR technologies, leveraging SNV-derived PAMs to enhance precision in gene editing.
QUICK START
Running AlPaCas is straightforward. Watch these videos for a quick start!
[Required] - Gene or Variants IDs
AlPaCas is thoughtfully designed to ensure a user-friendly experience with minimal input requirements. When using AlPaCas, you have two primary options to kickstart your analysis:
-
GeneSymbol/GeneID: If you intend to analyze all ClinVar-annotated Single Nucleotide Variants (SNVs) for a specific gene, all you need to provide is the GeneSymbol (HGNC nomenclature) or the GeneID.
-
VariationID: If your focus is on a single, or a few, specific variants annotated in ClinVar, simply enter the VariationIDs (VCV identifier) in a comma-separated list. Example: to analyze THIS variant with VCV000732689.3, insert 732689.
Check available genes HERE. This will help you identify the genes that can be examined using our tool.
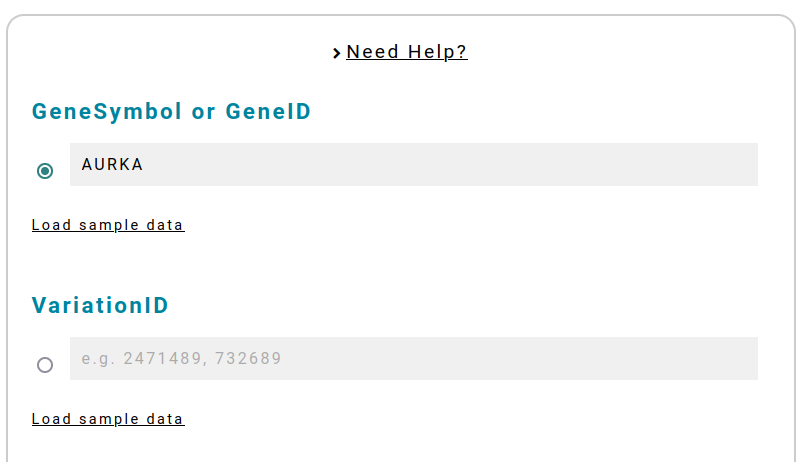
Click on Load sample data to run an example.
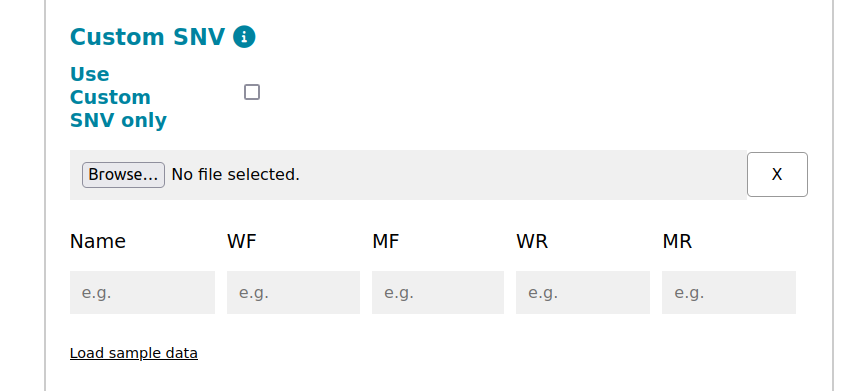
[Optional/Required] - Custom SNV
For SNVs not found in ClinVar, leverage the Custom SNV feature. Here's what you need to know:
-
Custom SNV only: check this option it if you intend to analyze only your SNVs. Otherwise, when adding your sequence of interest, you will also be prompted for input regarding the gene to be analyzed.
-
Name: Provide a unique identifier or "Name" for the custom mutation you want to analyze (see Section Special Characters and Names).
-
Sequences: Unlike ClinVar-annotated SNVs, AlPaCas relies on user-provided sequences when exploring non-annotated SNVs. You must provide sequences for the following: Wild-Type (WF): the original, unaltered sequence. Mutant (MF): the sequence with the SNV or mutation. Wild-Type Reverse (WR): the reverse complement of the original sequence. Mutant Reverse (MR): the reverse complement of the mutated sequence.
Sequence Length: The SNV must be the 26th caracther. Ensure that each of the provided sequences contains 25 nucleotides to the left and right of the mutation site (a total of 51 nucleotides).
-
Load from file: can be CSV files with the following column names: Name, WF, MF, WR, MR. Click on the button below to download a sample file or click on Load sample data to run an example.
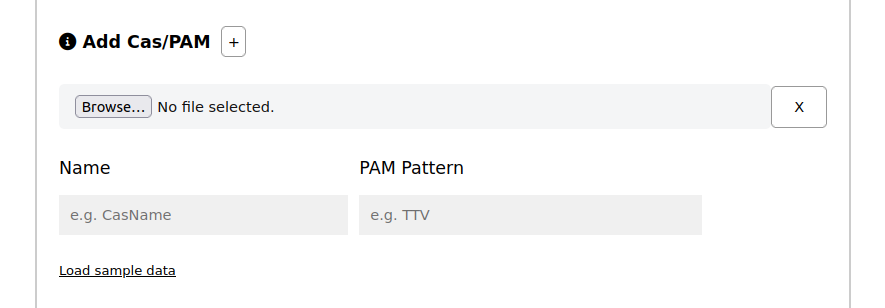
[Optional] - Add Cas/PAM
Analyzing Unlisted Cas Proteins
-
Name (see Section Special Characters and Names) and PAM Pattern: To analyze a Cas protein not found in our predefined list, you need to provide a unique "Name" for it along with the associated "PAM pattern" you wish to investigate.
-
Load from file: can be CSV files with the following column names: Name and PAM.
Click on the button below to download a sample file or click on Load sample data to run an example.
-
Limitation: When analyzing an unlisted Cas protein, please be aware that further structural analyses will not be available as them rely on the pre-processed 3D structure of well-annotated Cas proteins, which may not be present in the internal database. You can check the list of Cas proteins with pre-processed 3D structures here.
-
Usage note: When adding new PAM patterns, pay attention to the pattern as it will be influencing the stringency of the analysis. For a proper interpretation of the input, and related results, please refers to the section Interpretation of the results
Analyzing Unlisted Cas Proteins
-
Name (see Section Special Characters and Names) and PAM Pattern: To analyze a Cas protein not found in our predefined list, you need to provide a unique "Name" for it along with the associated "PAM pattern" you wish to investigate.
-
Load from file: can be CSV files with the following column names: Name and PAM. Click on the button below to download a sample file or click on Load sample data to run an example.
-
Limitation: When analyzing an unlisted Cas protein, please be aware that further structural analyses will not be available as them rely on the pre-processed 3D structure of well-annotated Cas proteins, which may not be present in the internal database. You can check the list of Cas proteins with pre-processed 3D structures here.
-
Usage note: When adding new PAM patterns, pay attention to the pattern as it will be influencing the stringency of the analysis. For a proper interpretation of the input, and related results, please refers to the section Interpretation of the results
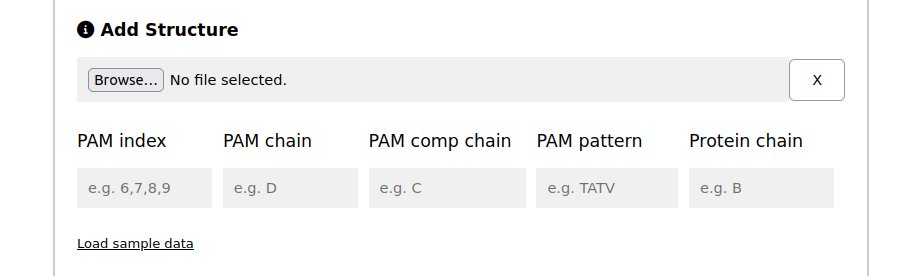
[Optional] - Add Structure
Analyzing Unlisted Cas Proteins with the structure
If you're analyzing unlisted Cas proteins, you might want to load your own structure for further analysis. In such cases, specific information about the structure is required.
-
PAM index: a comma-separated list of indices where to find the PAM sequence on the structure
-
PAM chain: the chain where the PAM is located
-
PAM comp chain: the chain where the PAM complementary sequence is located
-
PAM pattern: the PAM pattern associated with the imported structure
-
Protein chain: the chain of the protein itself
-
Load structure: load the structure in 'PDB' format.
As an example, load the structure provided here (button below) with the information provided by clicking on
Load sample data.
Analyzing Unlisted Cas Proteins with the structure
If you're analyzing unlisted Cas proteins, you might want to load your own structure for further analysis. In such cases, specific information about the structure is required.
-
PAM index: a comma-separated list of indices where to find the PAM sequence on the structure
-
PAM chain: the chain where the PAM is located
-
PAM comp chain: the chain where the PAM complementary sequence is located
-
PAM pattern: the PAM pattern associated with the imported structure
-
Protein chain: the chain of the protein itself
-
Load structure: load the structure in 'PDB' format. As an example, load the structure provided here (button below) with the information provided by clicking on Load sample data.
Explore the results
After running AlPaCas Finder, you can delve into the results via the dedicated results page. Each panel on this page provides detailed information about every variation associated with your input query. In the panels, each dedicated to an identified SNV, you will find:
-
A direct link to the ClinVar page related to the identified SNV.
-
The Clinical Significance as reported in ClinVar.
-
A Cas Targets Identified Widget to show the Cas proteins that can target the specific SNV-derived PAM. By scrolling through the widget, you can select different Cas proteins and explore how each Cas protein interacts with the sequence, which is dynamically highlighted. The region flanking the mutation that can be targeted by the selected Cas is highligthed in orange. Correspondingly, the sequence on the wild-type (WT) reference is highlighted in green, providing a clear contrast between the targeted region and the unaltered sequence.
-
A * symbol near a Cas highligths those SNV-derived PAM for which, according to the annotated PAM pattern, the mutation is being discriminated by a specific nucleotide.

-
Use our filtering tool to display only the SNVs that match specific criteria such as selected Cas targets and Clinical Significance. Browse the results by VariationID or refine your search by using the Filter by substring option, which will show SNVs with SNV-derived PAM sequences matching the inserted text.
Structural analysis
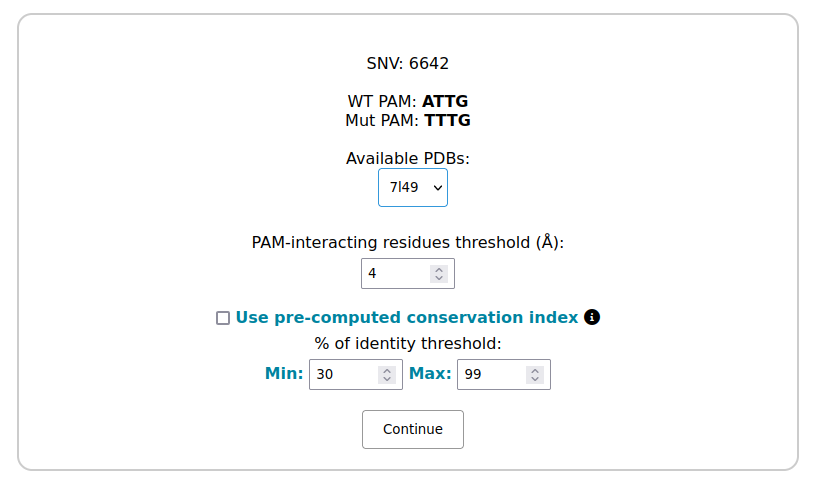
For each variant resulting from the previous search, a series of Cas targets capable of recognizing the PAM sequence generated by the SNV mutation are identified. If there are 3D structural details available for that specific protein case, it can be subjected to structural analysis by clicking the Run Structural Analysis button.
-
Available PDB: in this section, you can select the three-dimensional structure to undergo analysis. Each available structure is indicated by an alphanumeric PDB code.
-
PAM interacting residue threshold: define the range within which residues in contact with the PAM are taken into consideration.
% of identity threshold: This parameter allows you to define the range of identity within which homologous sequences will be selected. By aligning these sequences, evolutionary conservation scores are calculated and assigned to the previously selected residues.
-
Use pre-computed conservation index: by checking this box, the process will skip the evolutionary computation step, using instead values pre-computed within a range of % of identity 30-99
Explore Structural Analysis Results
-
Structural Analysis Summary: In this section, you can find a box containing a summary of the parameters previously provided as input.
-
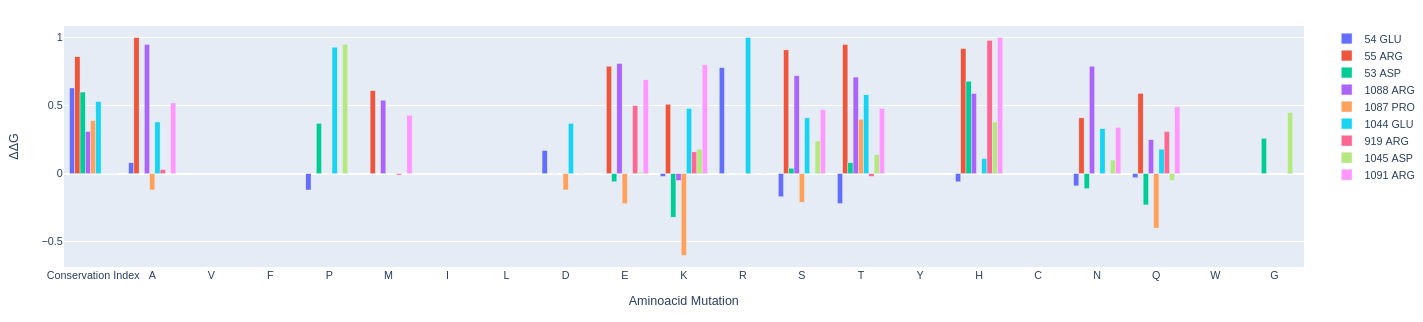
Interactive Table and viewer: The table displays the variation in ΔG interaction between the mutants and the wildtype protein. Therefore, structures displaying a negative ΔΔG demonstrate a better interaction energy with the nucleic acid (PAM). By clicking on the ΔΔG value shown in the table, the corresponding mutant is visualized in the molecular visualization widget, allowing you to view the mutant's structure.
- Conservation-index: The conservation index is a score that assesses how conserved a site is through an evolutionary analysis based on the alignment of homologous sequences. It varies between 0 and 1, where 0 indicates a poorly conserved site, while 1 indicates a highly conserved site. In the table, each site is highlighted with a color scale ranging from green to red, where green is associated with a low conservation index and red with a high conservation index
-
ΔΔG Graphic Visualization: The information displayed in the previously described graph is also presented in an interactive plot.
Download: Through the download buttons, you can save and retain the results of the structural analysis.
- Download table: Allows you to download the content of the table in CSV format.
- Download all results: Initiates the download of a compressed folder (.zip) containing the wild-type protein structure, structures of all mutants, and summary files.

Interpretation of the results
Interpreting the results generated by AlPaCas Finder requires consideration of potential ambiguities arising from factors such as the presence of multiple PAM patterns associated with the same Cas enzyme. In CRISPR-Cas systems, variations in PAM annotations and the flexibility of Cas enzymes can lead to diverse interpretations of PAM recognition. Consequently, users may encounter discrepancies or variations in the data presented. These differences can arise from variations in PAM annotations across different studies or from the inherent flexibility of Cas enzymes in recognizing PAM sequences. Such variations can impact the selection of the most suitable Cas enzyme for a given application, as different PAM patterns may exhibit varying degrees of specificity and efficiency. Therefore, users are encouraged to carefully evaluate and interpret the results in consideration of these potential variations to make informed decisions regarding their experimental designs or applications.
When faced with multiple Cas proteins that could potentially target the same SNV, how do I determine which one to select?
-
Is there ambiguity on the PAM pattern associated with a certain Cas? Check for less-preferred PAMs here
-
Is this SNV-derived PAM being discriminated with a specific nucleotide on the mutation? Check for * symbol near the SNV-derived PAM
-
How strong is the selectivity score for a given PAM pattern? Check it here
Such considerations must also be considered when adding new PAM patterns to the analysis, as these may influence the results
JobID
After initiating the process, you can retrieve it by referencing the JobID (please note that JobIDs expire on a monthly basis).
-
JobID: Search protocol job
-
S-JobID: Structural analysis job
Alternatives to retrieve your Job:
-
copy the JobID-URL and directly return to the page of the corresponding Job by pasting it into your browser's search bar
-
Bookmark the running page
-
Exploit the JobID search widget
Info tables
*PAM selectivity score: N = 0.25; V,H,D,B = 0.33; M,R,W,S,Y,K = 0.50; A,C,G,T = 1.00
Glossary
-
SNV: Single Nucleotide Variant
SNV-derived PAM: A SNV that creates a PAM solely in the mutant and not in the wild-type sequence.
- !! We define an SNV-derived PAM as the matching event between an SNV and a PAM pattern, when it is selective for the mutant allele. An SNV is any mutation that is being analyzed, when an SNV is targeted by a certain Cas in an allele-specific manner, it becomes an SNV-derived PAM.
WF: Wild-Type forward sequence
MF: Mutant forward sequence
WR: Wild-Type reverse sequence
MR: Mutant reverse sequence
Special characters and Names
Please avoid using these characters: !@#$%^&*()_+{}[]:;<>,.?~/- in the custom inputs, as they will be automatically replaced, impacting the expected name for your input. The name inserted in Custom SNV and Add Cas/PAM, or the file in Add Structure, is modified in order to avoid the presence of special characters and the presence of numbers as first characters in the string. Moreover, when a structure is loaded, the file name will also contain its PAM pattern. e.g. when loading '5b2t.pdb' file, its name will be 'S5b2tTATV.pdb'.
WORKFLOW & IMPLEMENTATION
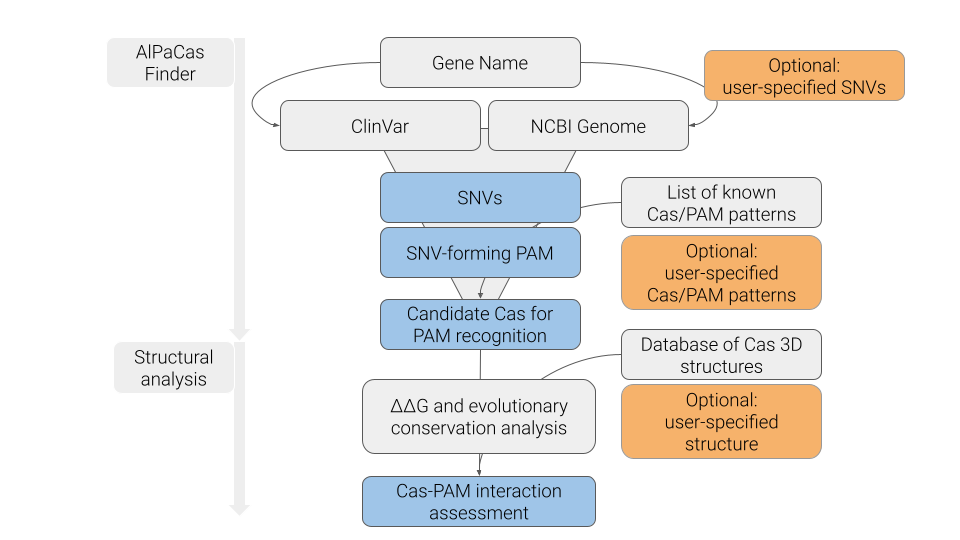
Here is a workflow scheme for AlPaCas. As explained in other sections, the input/methods of the analysis are shown in grey, while the optional inputs are highlighted in orange. The outputs are presented in light blue.
Databases interrogation: ClinVar entries and genomic sequences obtained from the NCBI have been adjusted for local interrogation through an SQL-structured database. Each locally adapted database is equipped with an automated monthly update method.
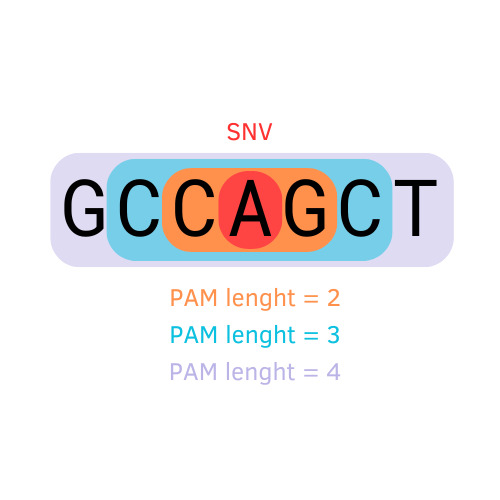
The Search protocol (aka AlPaCas Finder) is implemented in Python. By cross-referencing a source of SNVs with a compendium of established PAM patterns corresponding/linked to their respective Cas proteins, it identifies potential Cas proteins adept at recognizing the SNV-derived sequences. For every SNV, the protocol scans the sequences - both wild-type and mutated, as well as their forward and reverse orientations - flanking the site of mutation, utilising the list of PAM patterns. The extent of the nucleotides flanking the mutation that are considered for the screening depends on the length of the PAM pattern: see the image on the right
-
Structural Analysis Protocol: following the Search protocol, depending on the availability of the 3D structure (see info tables section) , the PAM-interacting residues are analyzed in terms of evolutionary conservation and ΔΔG. In essence, this step is designed not for predictive purposes but rather to establish the foundational guide for engineering Cas proteins specifically customized for PAM interaction.
- Evolutionary Analysis: the cas of interest undergoes a BLAST-search for homologoues sequences, which are going to be the input of the Multiple Sequence Alignment (MSA). In turns, the MSA is analyzed to asses a per-residue conservation index. In this way, each selected PAM-interacting residue is assigned to a conservation index that describes the degree of which a mutation at that site could lead to a detrimental effect on the protein.
- Substitutions Modelling and ΔΔG Assessment: selected residues undergo substitutions based on a BLOSUM62 substitution matrix, evaluated by making use of UniDesign, a computational protein design framework. It models the protein subjected to all possible substitutions and assesses the Cas-PAM interaction. Proteins exhibiting a negative ΔΔG are expected to demonstrate an improved ability to recognize the PAM.